Naukowcy z Instytutu Biologii Ewolucyjnej autorami nowego narzędzia bioinformatycznego do klasyfikacji sekwencji z danych metagenomowych
Doktorant mgr Michał Karlicki i student Stanisław Antonowicz wraz z dr hab. Anną Karnkowską z Instytutu Biologii Ewolucyjnej Wydziału Biologii UW są autorami artykułu naukowego, przedstawiającego nowe narzędzie oparte na metodach uczenia maszynowego do szybkiej i dokładnej klasyfikacji sekwencji eukariotycznych i prokariotycznych. Publikacja ukazała się na łamach czasopisma “Bioinformatics”.
Artykuł „Tiara: deep learning-based classification system for eukaryotic sequences” powstał w ramach badań finansowanych z EMBO Installation Grant a kierowanych przez dr hab. Annę Karnkowską.
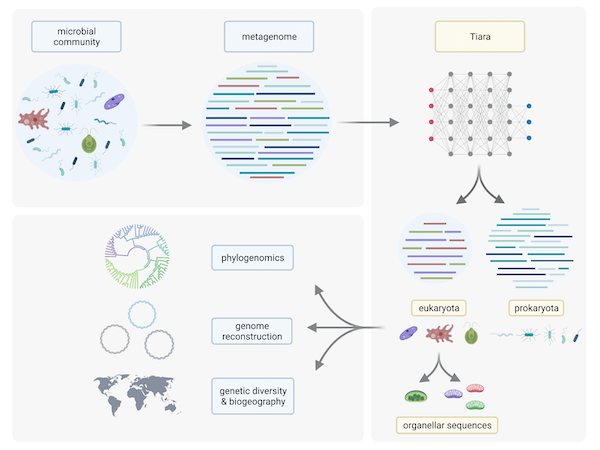
Wraz z rosnącą liczbą danych metagenomowych pochodzących z sekwencjonowania wysokoprzepustowego mikroorganizmów w różnych środowiskach poznajemy coraz lepiej ich różnorodność i funkcje. Dużym wyzwaniem jest jednak odzyskiwanie i prawidłowa klasyfikacja sekwencji eukariotycznych. Celem zespołu dr Karnkowskiej było przygotowanie narzędzia, które dokładnie i szybko identyfikuje sekwencje eukariotyczne oraz dzieli je na sekwencje pochodzące z genomu jądrowego, mitochondrialnego i chloroplastowego, co jest niezbędnym krokiem w kierunku lepszego zrozumienia różnorodności eukariotów.
Opracowane przez zespół narzędzie o nazwie Tiara opiera się na uczeniu maszynowym i służy przede wszystkim do identyfikacji sekwencji eukariotycznych w danych metagenomowych. Zastosowano w nim dwuetapowy proces klasyfikacji, co umożliwia identyfikację sekwencji jądrowych i organellarnych, a następnie dzieli sekwencje organellarne na plastydowe i mitochondrialne. Przeprowadzone testy wykazały, że Tiara dokładniej klasyfikuje sekwencje eukariotyczne niż inne dostępne narzędzia i jest jedynym programem, który identyfikuje sekwencje organellarne. Tiara dobrze radzi sobie zarówno z danymi pochodzącymi z mikrobiomów organizmów eukariotycznych, jak i z dużymi i złożonymi zestawami danych obejmującymi różnorodność mikroorganizmów w oceanach.
Narzędzie i wskazówki dotyczące instalacji oraz użytkowania znajdują się w ogólnodostępnym repozytorium GitHub. Zapraszamy do korzystania!